GlobDB release 232 methods
Dereplication of the underlying databases
The GlobDB release 232 integrates datasets that provide a dereplicated set of genomes, using a 95 % average nucleotide identity (ANI) dereplication criterion, and datasets where a dereplication step (also at 95% ANI using Galah v0.4.2) is performed during the building of the GlobDB. Since the underlying data for each database is distinct, there may be centroids between datasets that belong to the same species by the operational definition of 95 % ANi similarity, but between them represent a cluster of genomes that extends beyond that operational species boundary. Thus it is possible that the genomes that two centroids represent wouldn't all be classified to the same species if all data was processed together.
The GlobDB therefore conservatively dereplicates the underlying (already dereplicated) datasets at 96 % ANI over 50 % aligned fractions (AF). We have chosen these cutoffs after inspection of ANI histograms and crossplots of ANI vs AF values (see examples below), showing a clear ANI gap between the dereplicated datasets starting at approximately 96% ANI.
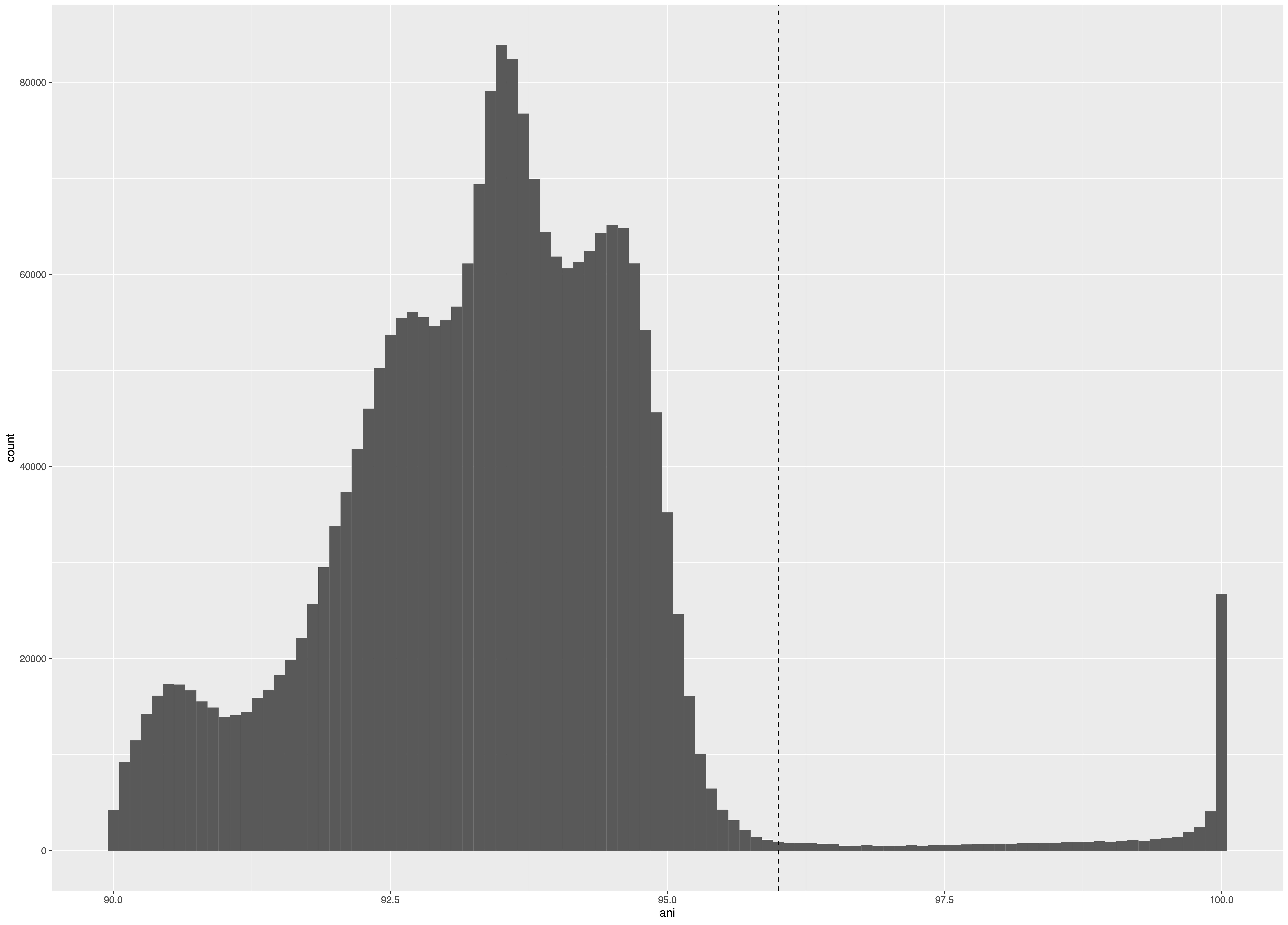
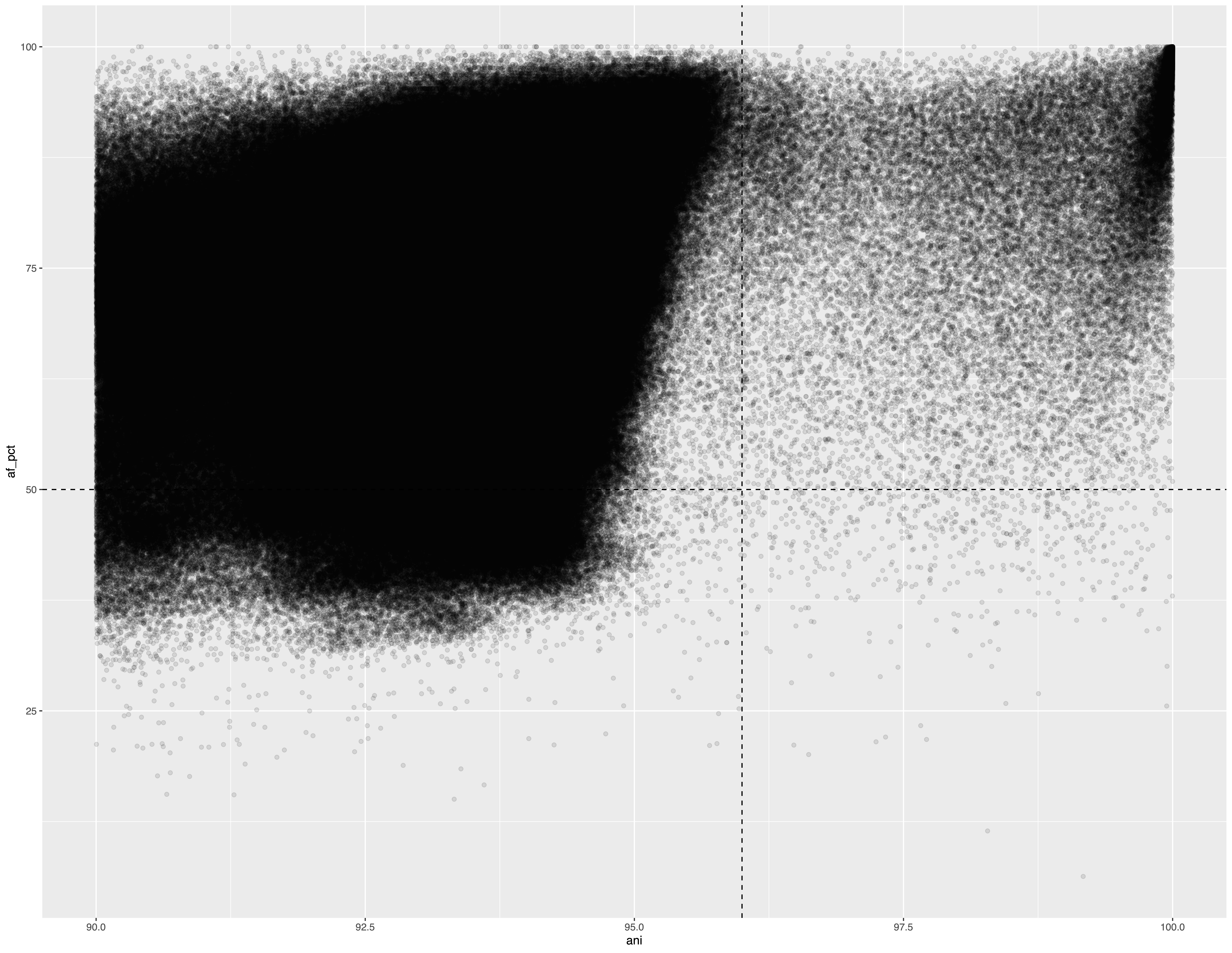
Left: Histogram showing the pairwise ANI values of the GTDB (release 226) and the mOTU 4.0 species representatives. Right: crossplot of ANI and aligned fractions (AF) of the GTDB (release 226) and the mOTU 4.0 species representatives. Dashed lines indicate the cutoffs for inclusion in the GlobDB at 96% ANI and 50% AF.
The final GlobDB dataset is as follows
- A MAG dataset that has not been deposited in INSDC databases is downloaded.
- If a species dereplicated set is defined by the authors and available, this set is used.
- If no species dereplicated set is defined, the MAGs are dereplicated at 95% ANI using Galah 0.4.2, using checkM2 quality scores.
- The dereplicated dataset is then dereplicated against the other non-INSDC datasets using the reference mode of Galah at 96% ANI, and checkM2 quality scores.
- This creates a dereplicated "non-INSDC" dataset to which further datasets can be added.
- Finally, after a new GTDB release, this dereplicated "non-INSDC" dataset is dereplicated against the GTDB species representatives using the reference mode of Galah at 96% ANI, and checkM2 quality scores.
To ascertain that the GlobDB contains all GTDB species representatives, the GTDB species representatives are given priority over the genomes in the "non-INSDC" dataset, regardless of CheckM2 quality assessment.
The complete final dataset was then assessed for quality using CheckM2 and anvi'o. There were four reasons for exclusion of genomes at this stage, in this order:
1) genomes that failed the annotation process
2) genomes with an anvi'o domain designation EUKARYA
3) genomes with a total length over 20M bp
4) genomes where "CheckM2 completeness - 5*CheckM2 contamination" evaluates to less than 50
To ascertain that the GlobDB contains all GTDB species representatives, GTDB species representatives that do not meet the standard set in step 4 are still included in the final dataset.
For convenience in later use, the names of the genomes in the all datasets except the GTDB, MGnify, and MRGM/HRGM2 are standardized to GlobDB identifiers that represent their origin and are propagated throughout the GlobDB resources. Dictionary files to relate the GlobDB IDs back to the identifiers from the original publications are available in the Downloads page.
Generation of anvi'o databases and annotation
After dereplication, the resulting genome fasta files are turned into anvi'o contigs databases and basic annotation is performed.
First, contig names are standardized using anvi'o with anvi-script-reformat-fasta and the options: --seq-type NT # specifies the sequence type--simplify-names # standardizes the contig name format--overwrite-input # overwrites the source file--prefix <GlobDB_ID> # prepends the GlobDB ID to the contig name
After this, contigs databases are created using anvi-gen-contigs-database, using the GlobDB ID as database name (-n flag). Basic annotations on these contigs databases are run using HMMs integrated with anvi'o. Information on the sources of HMMs that are included in anvi'o can be found here. Addditionally, KEGG, COG, Pfam and CAZy annotations are performed. Please see the links in the file descriptions on the Downloads page for more information on the anvi'o implementation of these annotations. One specific thing to note is that for KEGG annotations, anvi'o employs a heuristic to add (likely) true positive hits that were missed during the first pass, described in more detail here.
These contigs databases are then populated with annotations using the following commands:anvi-run-hmms # annotates rRNA genes, marker genes, and tRNAs anvi-run-kegg-kofams # annotates coding regions with kegg kofamsanvi-run-pfams # annotates coding regions with pfamsanvi-run-cazymes # annotates coding regions with dbCAN2 CAzymesanvi-run-ncbi-cogs # annotates coding regions with COGs (2020 update)
Next, protein fasta files and gff files for KEGG, COG and Pfam annotations are exported from the contigs databases
These files can also be generated from the anni'o databases directly using the anvi-get-sequences-for-gene-calls command with different flags.
# protein fasta--get-aa-sequences --wrap 0 --defline-format {contigs_db_project_name}___{gene_caller_id} # gff with COG annotation--export-gff3 --annotation-source COG20_FUNCTION
Establishing the GlobDB Taxonomy
De novo genome trees for bacterial and archaeal genomes from novel species were generated using GTDB-tk v2.4.0 de_novo_wf (Chaumeil et al. 2022). Novel clades were identified through a custom algorithm based on relative evolutionary divergence (RED), similar to that described in Supplementary Figure S1 from Chaumeil et al. 2020. Firstly, RED values were calculated by PhyloRank v0.1.12 using the de novo genome trees from GTDB-tk. Next, the RED values and genome trees were processed using a custom script (see name_clades.py here). Genomes were sequentially assigned to either pre-existing or novel clades, ordered by genome quality. For each genome, this involved traveling up the tree until a node was reached within the RED-bounds of a new taxa level (e.g. phylum, class, order, etc.) for which the genome has not yet been assigned. Parents and children of the node were checked to find if any were closer to the median RED of that taxa level (from GTDB R226) than the current node. If the current node was the closest node, it is annotated as a new clade, with the current genome being assigned as the genome representative. This continued until a node was reached that had an offspring from GTDB (or until reaching a previously labelled node). The remaining taxonomy was assigned based on the median taxonomy of the GTDB offspring from that node. A snakemake workflow implementing the above can be found in this github repository.